Page 9, equation (2.5): there is a minus sign missing in the exponent before the alpha in the Morse potential.
Page 55, equation (3.12): the subscript on the first two wave functions should be nj instead of ni:
![]()
Page 62, equation (3.35): the subscript on the first J operator should be j instead of i:
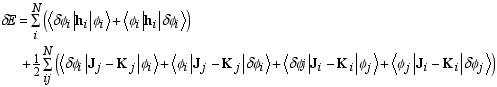
Page 66, equation (3.54): the subscripts on the last D element should be gamma,beta and not delta,beta.
Page 78, line 3: cubic should be quartic: The only limitation is the quartic (M4) growth of the memory requirement ...
Page 124: equation (4.34), subscript on W after the summation should be: 2n+1-k-l not 2n+1-k-1 (i.e. the last character should be an L and not a 1 (one)).
Page 125: equation (4.35), first equation: psii should be psi1.
Page 141: equation (4.67), last equation, the operator r13 should be r34.
Page 149: reference 41 should be: H. Koch, A. S. de Meras, T. Helgaker and O. Christiansen, J. Chem. Phys. 104 (1996) 4157.
Page 178, equation (6.1): a minus sign is missing on the right hand side in the equation for Ene.
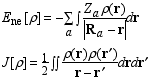
Page 184, equation (6.22): In the denominator, the term beta4x3 should be beta4x4.
Page 186, equation (6.28), third line: In the definition of delta, the exponent of the first rho should be -1/3, and not +1/3, i.e. delta = crho-1/3.
Page 224-225: P. Popelier has pointed out that the discussion of AIM charges in section 9.3, and especially Figure 9.3, may be misleading.
For a given atomic basin, the electron density as seen from the outside gives raise to an electrostatic potential, which may be expanded in terms of electrical moments. The electron density may be integrated and combined with the nuclear charge to give a net charge, the first moment, which is the AIM "atomic charge". Higher order atomic moments (dipole, quadrupole etc.) may similarly be defined by suitable integrations of the electron density.
If the net charges are taken as nuclear centered (analogous to partial charges for force field methods) they do not reproduce the molecular dipole, quadrupole etc. moments, nor do they yield a good representation of the electrostatic potential. They are therefore not suitable for transferring to a force field environment for modelling purposes.
If, however, the dipole moments of the atomic basins are also considered, the total molecular dipole moment is reproduced, and similarly for higher order moments. The dipole moment of CO, for example, is close to zero (0.122 D), despite calculated AIM charges of +/- 1.1. The large charge transfer is almost exactly canceled by compensating atomic dipoles.
C. L. Perrin (ref. 14) has attributed the large AIM charges to orbital effects, as illustrated in Figure 9.3. C. Gatti and P. Fantucci (J. Phys. Chem. 97 (1993) 11677), however, have shown that Perrins arguments are only valid if orbital overlaps are neglected. For ab initio wave functions this is not a valid approximation.
Page 243, equation (10.33), middle equation: The derivative of the CI wave function on the right hand side is not with respect to C but with respect to c (i.e. not all parameters, just the MO-coefficients), i.e. dpsiCI/dC --> dpsiCI/dc.
Page 303, equation (12.22), the formula for Srot: The factor 1/2 should only go with the 3 on the inside of the parenthesis, i.e. Srot = R[3/2 + ln(...)].
Page 312, equations (13.13) and (13.14), and text between: The FG matrix is not the product of the F and G matrices, but should be taken as containing the product of the matrix elements, i.e. (FG)ij = FijGij.
Page 313, just below equations (13.18): The ra,b,c vectors are not the eigenvectors of the inertia matrix (which are of length 3), but vectors constructed from the eigenvalues of the inertia matrix and the atomic coordinates (i.e. vectors of length 3N). See ref 13.1 for details.
Page 323, line 11: The 3N-1, 3N-2 and 3N-3 should be N-1, N-2 and N-3, i.e. For a simple acyclic system these may be chosen as N-1 distances, N-2 angles and N-3 torsional angles, ...
Page 374, equation (16.6): There is a factor of 1/Nstates missing in the numerator of the second and third equations, since the "1" is being replaced by 1/Nstates times a sum over eEi/kTe-Ei/kT. This constant factor can be seperated out and just corresponds to a shift in the zero point for the Helmholtz free energy.
Page 397, equation (16.52), third equation: the a2ij should be aij:
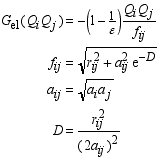
Page 408, equation (B.9), fourth equation: the right hand E'0 should not have a prime, and should be italic, i.e. E0.
Page xiv, line 26: 850 should be 850 amu.
Page 7, line 2: right parenthesis is missing: ... see Ref. 3).
Page 29, line 13: word missing: ... for comparing with experimental data.
Page 31, line 11: where should be whether: ... depending on whether the fitting is done ...
Page 31, line 21: atoms should be atom: ... i.e. more atom types must be used.
Page 32, line 3: right parenthesis is missing: ... can be found in Ref. 22).
Page 41, footnote: Gunsterenm should be Gunsteren.
Page 45, line 25: word missing: ... one of the largest molecule to have been subjected to ...
Page 54, equation (3.3): subscript on Vee should not be italic, i.e.: Vee --> Vee
Page 69, just above eqs. (3.62): witten should be written, and just after the parenthesis, follow should be followed.
Page 71, line 28: calculation should be calculate
Page 74, line 5 from the bottom, excess word that: ..., i.e. sufficiently near the minimum it converges very fast.
Page 98, line 16, missing word is: This is not the same as saying....
Page 102, equation (4.2): the subscript on the first Slater determinant should be HF instead of SCF
Page 127, equation (4.40), third equation, and equation (4.41): the subscript on the Vee operators should not be bold.
Page 152, line 11 and 12: clarifying that it is sets of p-functions: ... and two sets of p-functions (2p and 2p') for first row elements, and six s-functions and four sets of p-functions for second row elements..
Page 160, line 6 from the bottom: the TZ type DH basis is a (12s9p) primitive basis set contracted to [6s5p] (not (13s9p) --> [6s4p]) with the contractions 6,3,1,1,1,1 for the s-functions (note doubling of one function) and 4,2,1,1,1 for the p-functions.
Page 162, line 27: addtional should be additional.
Page 165, line 9: word missing: ... may be defined ....
Page 175, reference 18: the volume 3 should be 1.
Page 176, reference 24: first two authors should be K.A. Peterson, A.K. Wilson.
Page 176, reference 28: J. A. Curtiss should be L. A. Curtiss.
Page 181, line 1: chose should be choose.
Page 181, equations (6.10) and (6.11): the KS subscript on the h operator should not be italic, hKS --> hKS .
Page 185, equation (6.24): an epsilon is missing on the left hand side of the second equation defining the Becke energy correction.
Page 188, line 10: fiiting should be fitting.
Page 193, reference 14: J. D. Perdew should be J. P. Perdew.
Page 193, reference 15: Phys. Rev. B should be Phys. Rev. A..
Page 193, reference 24: the year should be (1993).
Page 216, Table 8.1, header and table: the nabla sign should be a delta sign.
Page 218, line 10: there should not be commas seperating the alphas in the subscript of D and S.
Page 219, line 30: the charge +10 should be +8, i.e. .., in which case the electrons are not associated with any nuclei at all, i.e. atomic charges of +1 and +8!
Page 227, line 19: an should be a ... optimizing the expectation value of a two-eletron operator....
Page 227, eqs. (9.21): right ket should be larger.
Page 228, eqs. (9.23): right ket is missing.
Page 229, just above eqs. (9.29): the * denoting complex conjugation should be a superscript, i.e. psi*psi.
Page 229, eqs. (9.29): the integration should also be over r', i.e. drk+1...drNdr'k+1...dr'N.
Page 230, eqs. (9.30): the last subscript should be n+2 instead of n+1.
Page 237, eqs. (10.6) is labelled as (19.6).
Page 300, eqs. (12.14): summation index i on x is missing in the top right hand corner element.
Page 398, reference 18: P. M. Ceperley should be D. M. Ceperley.
Page 409, equation (B.10): the right hand E'0 should be italic, i.e. E'0.
Trygve Helgaker, Poul Jørgensen and Jeppe Olsen have a new text book out which treats all standard Ab Initio methods (HF, MCSCF, CI, CC, PT, basis sets) in a very comprehensive way: Molecular Electronic-Structure Theory.
Jonathan M. Goodman has a related book: "Chemical Applications of Molecular Modelling".
"Exploring aspects of computational chemistry: concepts and exercises" is a related book in the same area.
"Encyclopedia of Computational Chemistry" contains a wealth of useful information.
For some new developments in inclusion of polarization effects (i.e. many-body electrostatics) in force fields, see J. L. Banks, G. A. Kaminski, R. Zhou, D. T. Mainz, B. J. Berne, R. A. Friesner, J. Chem. Phys. 110 (1999) 741.
An updated description of the GROMOS force field/program has been published: J. Phys. Chem. A 103 (1999) 3596.
Brian Sutcliffe has a review on the separation of electronic and nuclear motions (Born-Oppenheimer approximation) in Adv. Chem. Phys. 114 (2000) 1-123.
For a comparison of different linear scaling methods for achieving SCF solutions, see A.D. Daniels, G. E. Scuseria J. Chem. Phys.110 (1999) 1321.
Matt Challacombe has a web page on linear scaling methods.
The use of fractional occupation numbers for improving SCF convergence: A. D. Rabuck, G. E. Scuseria J. Chem. Phys. 110 (1999) 695.
M. W. Schmidt and M. S. Gordon have a nice review of MCSCF methods in Annu. Rev. Phys. Chem. 49 (1998) 233.
C. D. Sherrill and H. F. Schaefer III have a nice review of CI methods in Adv. Quant. Chem. 34 (1999) 143.
The T1-diagnostic for evaluating the quality of a CCSD wave function has been shown to be dependent on the number of electrons, and two new diagnostics for MP2 and CCSD, denoted D1(MP2) and D1(CCSD) have been defined: C. L. Janssen, I. M. B. Nielsen, Chem. Phys. Lett. 290 (1998) 423.
Two additional tests for wave function quality has been proposed: D2(MP2) and D2(CCSD): I. M. B. Nielsen, C. L. Janssen Chem. Phys. Lett. 310 (1999) 568.
P. Y. Ayala and G. E. Scuseria have developed CC and MP2 methods which displays linear scaling with system size, J. Chem. Phys. 111 (1999) 8330,
M. Schutz, G. Hetzer and H.-J. Werner have developed a local MP2 method which displays near linear scaling with system size, J. Chem. Phys. 111 (1999) 5691.
The use of isodesmic reactions for improving the performance of G2 extrapolation procedures: J. Chem. Phys. 106 (1997) 6764.
A new Gaussian-3 composite model has been proposed which reduces the MAD error to 1.0 kcal/mol (from 1.5 kcal/mol with the G2 method): L. A. Curtiss, K. Raghavachari, P. C. Redfern, V. Rassolov, J. A. Pople, J. Chem. Phys. 109 (1998) 7764.
Martin and Oliveira have proposed two new composite models (W1 and W2), which are able to calculate heats of formation with MAD errors of 0.30 and 0.23 kcal/mol, respectively: J. Chem. Phys. 111 (1999) 1843.
The performance of various CBS extrapolation procedures: J. Chem. Phys. 108 (1998) 692.
The exponential convergence of the Hartree-Fock energy with respect to basis set size has been explicitly demonstrated: J. Chem. Phys. 110 (1999) 6601. and Theo. Chim. Acc. 104 (2000) 484
The EMSL Gaussian basis set order form is a good place for obtaining a variety of basis sets.
Additional reference for deriving DFT functionals from ab initio data: J. Chem. Phys. 108 (1998) 2545.
R. Stowasser and R. Hoffmann have some thoughts on the importance of DFT orbitals and eigenvalues, J. Am. Chem. Soc. 121 (1999) 3414.
Dyall and van Lenthe have generalized the ZORA and FORA methods to IORA (Infinite Order Regular Approximation): J. Chem. Phys. 111 (1999) 1366.
The Darwin correction has been shown to converge as L-1 with respect to the highest angular momentum functions in the basis set. This is significantly slower than the correlation energy in non-relativistic theory: A. Halkier, T. Helgaker, W. Klopper, J. Olsen Chem. Phys. Lett. 319 (2000) 287.
P. Popelier has a new book on the AIM concept: Atoms in Molecules Pearson Education 1999.
A review of methods for calculating electrical non-linear properties: H. A. Kurtz, D. S. Dudis, Rev. Comp. Chem. 12 (1998) 241.
A review of methods for calculating NMR parameters: T. Helgaker, M. Jaszuski, K. Ruud, Chem. Rev. 99 (1999) 293.
Additional reference for CCSDT and CCSD(T) vibrational frequencies for O3 with cc-pVXZ basis sets (X = 2, 3, 4, 5): J. D. Watts, R. J. Bartlett, J. Chem. Phys. 108 (1998) 2511.
The NIST group has a list over molecules which are problematic at some particular levels of theory.
Other good books on reaction dynamics and transition state theory are:
R.G. Gilbert, S.C. Smith "Theory of Unimolecular and Recombination Reactions" Blackwells, 1989.
J. I. Steinfeld, J. S. Francisco, W. L. Hase "Chemical Kinetics and Dynamics" Prentice Hall, 1999 (second eds.).
B. K. Carpenter has a nice review of reactions displaying non-statistical product distribution: Angew. Chem. Int. Ed. Eng. 37 (1998) 3340. He also has a web-site with more information.
Joseph W. Ochterski has a document describing in detail how frequencies are calculated in Gaussian.
A new method for global optimization based on deformation of the original surface, called Bad Derivative Method, has been proposed: I. Andricioaei, J. E. Straub, J. Comp. Chem. 19 (1998) 1445.
Baker and Pulay have shown that inverse distance coordinates in combination with delocalized internal coordinates are very efficient for optimization of molecular clusters: J. Comp. Chem. 21 (2000) 69.
A good review of methods for calculating free energies: H. Meirovitch, Rev. Comp. Chem. 12 (1998) 1.
C. J. Cramer and D. G. Truhlar have a review on continuum solvation models: Chem. Rev. 99 (1999) 2161.
D. Frenkel and B. Smit have a web page in connection with their "Understanding Molecular Simulation: From Algorithms to Applications" book.