Current Research Interests (woefully out of date)
The current research topics in the Peterson group can be separated into four main categories
- Gaussian basis sets for molecular calculations (correlation consistent basis sets)
- Ab initio calculations of the thermodynamics and spectroscopy of small molecular clusters
- Accurate electronic structure calculations of ground and electronically-excited potential energy surfaces for atmospherically important molecules
- Accurate calculation of multi-dimensional potential energy surfaces for weakly-bound molecules and complexes
Gaussian basis sets for molecular calculations (correlation consistent basis sets)
Current and recent projects (this link for all available downloads):
- New families of correlation consistent basis sets, cc-pVnZ-PP, for use with accurate relativistic pseudopotentials
- post-d groups 13-18 [J. Chem. Phys. 119, 11099 (2003), ibid, 1113 (2003)]. Available on EMSL website.
- Groups 11 & 12 transition metals [Theor. Chem. Acc. 114, 283 (2005)]
- 2nd and 3rd row transition metals
- Effect of tight f functions for post-d main group elements, cc-pV(n+f)Z-PP
- Basis sets for explicitly correlated methods, MP2-F12
- 1st and 2nd row atoms, cc-pVnZ-F12 [J. Chem. Phys. 128, 084102 (2008)].
- alkali and alkaline earth metals (Li, Be, Na, Mg)
- F12 basis sets for core correlation effects, cc-pCVnZ-F12
- F12 basis sets with relativistic pseudopotentials, post-d elements Ga-Rn, cc-pVnZ-PP-F12
- Fitting basis sets, RI sets for F12
- 1st and 2nd row atoms, Optimized RI sets for cc-pVnZ-F12 and aug-cc-pVnZ
- DF and RI fitting basis sets for post-d main group elements and cc-pVnZ-PP orbital basis sets
- All-electron correlation consistent basis sets for the transition metals, nonrelativistic and DK-relativistic
- 3d-metals [J. Chem. Phys. 123, 064107 (2005)]. Available here and on EMSL website
- cc-pVTZ-DK sets available for 2nd and 3rd row metals (see link above)
- Basis sets for core-valence correlation effects
- 1st and 2nd row atoms, cc-pCVnZ and cc-pwCVnZ [J. Chem. Phys. 117, 10548 (2002).]
- transition metals - 1st row metals, 2nd row metals, groups 11&12
- post-d groups 13-18 with cc-pwCVnZ-PP basis sets
This work is supported by the U.S. National Science Foundation (CHE-0111282 and CHE-0723997 to KAP) and the U.S. Department of Energy, Division of Chemical Sciences, Basic Energy Sciences (through Pacific Northwest National Laboratory, Molecular Theory Group)
Current correlation consistent basis set bibliography
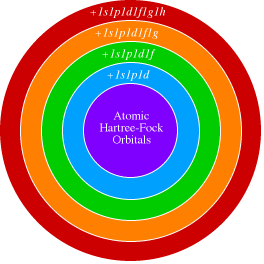 Correlation consistent basis sets are built up by adding shells of functions to a core set of atomic
Hartree-Fock functions. Each function in a shell contributes very similar amounts of correlation energy in an
atomic calculation.
Correlation consistent basis sets are built up by adding shells of functions to a core set of atomic
Hartree-Fock functions. Each function in a shell contributes very similar amounts of correlation energy in an
atomic calculation.
For the 1st and 2nd row atoms, the cc-pVDZ (correlation consistent-polarized valence double zeta) basis
set adds 1s, 1p, and 1d function. The cc-pVTZ set adds another s, p, d, and an f function, etc.
Various augmentations to these base sets have also been developed. These include the addition of diffuse
functions to better describe anions and weakly interacting molecules (aug-cc-pVnZ), as well as special basis
sets designed for describing the effects of correlating the core electrons (cc-pCVnZ and cc-pwCVnZ).
Ab initio calculations of the thermodynamics and spectroscopy of small molecular clusters
Current projects in this area include:
- Small transition metal clusters involving Group 11 and 12 elements
- Interactions involving sulfuric acid and water: H2SO4(H2O)n, HSO4-(H2O)n, SO4-2(H2O)n
- Binding enthalpies and structures of chlorinated hydrocarbon clusters: CCl4-CCl4, CCl4-H2O, CHCl3-CHCl3, etc.
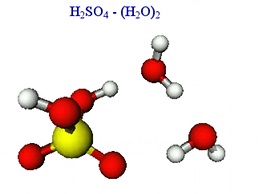 The picture to the right depicts one of several stable conformers of two water molecules interacting with
sulfuric acid. Each water molecule has a binding energy of about 12-13 kcal/mol as computed at the
MP2/aug-cc-pVDZ+d level of theory. Notice how each water molecule is both a proton acceptor and donor.
The picture to the right depicts one of several stable conformers of two water molecules interacting with
sulfuric acid. Each water molecule has a binding energy of about 12-13 kcal/mol as computed at the
MP2/aug-cc-pVDZ+d level of theory. Notice how each water molecule is both a proton acceptor and donor.
Accurate electronic structure calculations of ground and electronically-excited potential energy surfaces for atmospherically important molecules
Current and recent projects in this area include:
- Potential energy surfaces for reactions of mercury with reactive halogens, e.g., Hg + {Br2, BrCl, BrO}
- Chemically-accurate thermochemistry for Hg- and Cd-containing species
- Electronic spectra of hydrogen peroxide-water complexes (with J. Francisco, Purdue Univ.)
- Potential energy surfaces for the O(1D, 3P) + HCl reactions including spin-orbit couplings
- Potential energy surfaces of HOBr for unimolecular decomposition and the photodissociation process HOBr + hv → OH + Br
- Spin-orbit effects in atoms and small molecules: {F, Cl, Br}, BrCl, BrO, and HOBr
- Accurate prediction of rotational-vibration spectra of OClO, OBrO, HOCl, and HOBr from accurate 3-D ab initio potential energy functions.
- Large-scale potential energy surfaces of HOCl for describing vibrational energy transfer leading to OH + Cl
This material is based upon work supported by the National Science Foundation under Grant Nos. CHE-9501262 and CHE-0111282
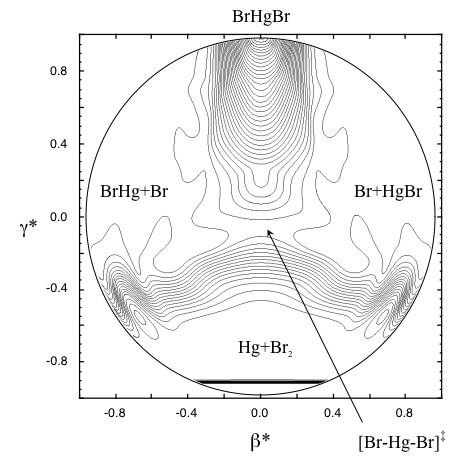
The global potential energy surface for the reaction of HgBr with Br features 3 reaction channels:
Recombination: HgBr + Br → HgBr2
Abstraction: HgBr + Br → Hg + Br2
Exchange: HgBr + Br → BrHg + Br
At 298K, the thermal rates are calculated to be fast and nearly equal for all 3 product channels. Oxidation of Hg by Br2 is calculated to be very slow at this temperature.
N.B. Balabanov, B.C. Shepler, and K.A. Peterson, J. Phys. Chem. A, submitted (6/05).
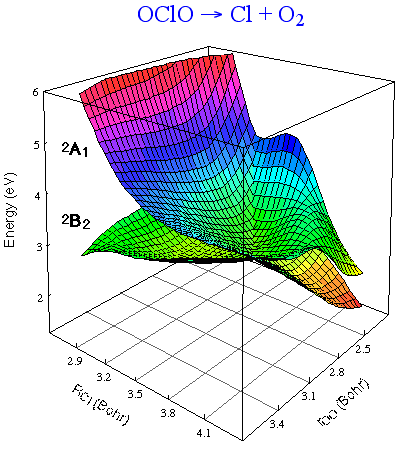
Photodissociation of OClO:
In this photodissociation two electronic states of the O2 product are observed: the ground and first excited states. Using large configuration interaction wave functions, 2-D portions of the exit channel were calculated to produce the figure at the right. The 2B2surface produces a1Δ O2, while the 2A1 surface produces X3Σ- O2. The lateness of the crossing suggests that more a1Δ O2 will be formed, which is consistent with experiment.
K.A. Peterson and H.-J. Werner, J. Chem. Phys. 105, 9823 (1996).
Accurate calculation of multi-dimensional potential energy surfaces for weakly-bound molecules and complexes
Current projects:
- Accurate He-CO interaction surfaces using CCSD(T) functions and explicit extrapolations to the basis set limit using a series of correlation consistent basis sets (with G. McBane, Grand Valley St. Univ.)
- K.A. Peterson and G.C. McBane, "A hierarchical family of three dimensional potential energy surfaces for He-CO", J. Chem. Phys., accepted (5/05).
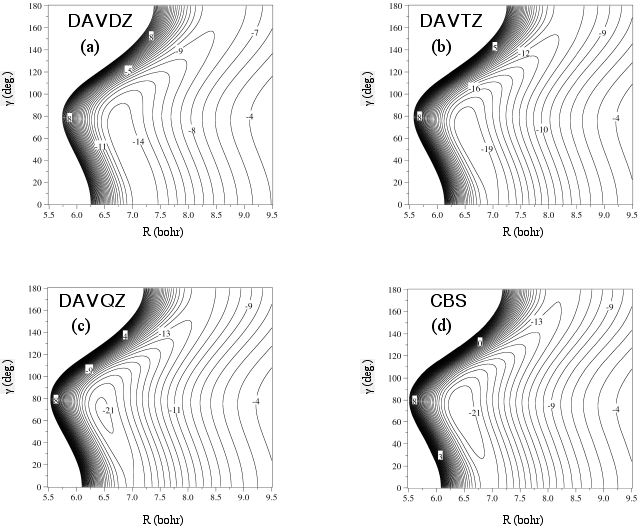
Potential energy contours (in cm-1) as a function of basis set at the CCSD(T) level of theory for the He-CO van der Waals complex.
R is measured from the He atom to the center of mass of CO (fixed at re=2.1322 bohr). Gamma=0 corresponds to C-O--He.